Regulació del software com a medical device: classe de risc i fases del procés
Disponible en VÍDEO! Roadmap regulatori necessari per configurar, validar i llançar al mercat un software com a medical device (MD) en el 10è Breakfast&Learn de la Xarxa TECSAM. De la mà de Dominique Monferrer, Medical Device Director d’Asphalion, coneixeràs la classificació del software en funció de la classe de risc i les fases del procés regulatori. No t’ho perdis!
La classificació del software en funció de la seva classe de risc (I, IIA, IIB o III) és un dels punts centrals a definir a la fase inicial de la creació d’aquest tipus de productes sanitaris
El programari com a medical device es classifica, per defecte, en risc IIA, cosa que incideix en el procés regulatori d’aquests productes sanitaris i precisa la implicació d’un organisme notificat per dur a terme la validació i la sortida al mercat. Aquesta ha estat una de les idees centrals del 10è Breakfast&Learn de la Xarxa TECSAM.
Aquesta nova cita del B&L, sota el nom «Regulació del software com a medical device», ha anat a càrrec de Dominique Monferrer, Director de Medical Device d’Asphalion, qui ha traçat els passos que tot investigador o investigadora ha de dur a terme per acreditar el seu software, des de la seva conceptualització i disseny fins a la seva validació clínica i la comercialització posterior.
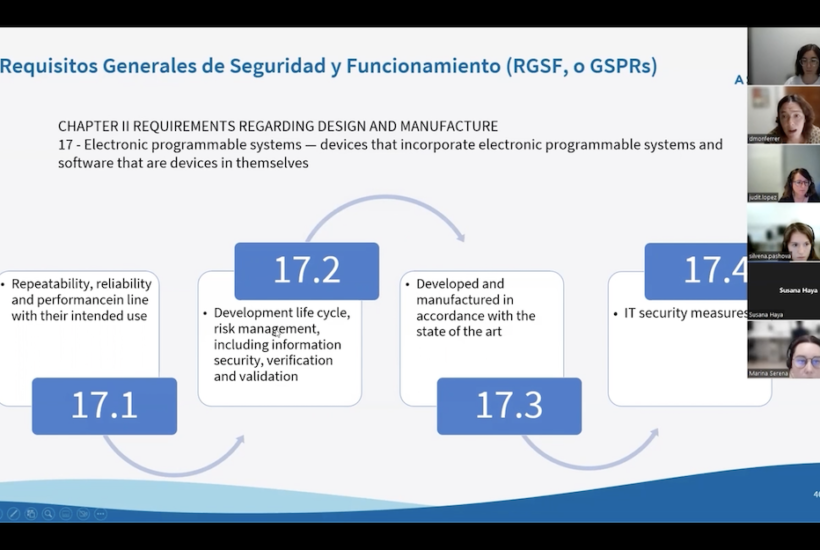
Un dels criteris imprescindibles a tenir en compte tant a la fase inicial de configuració del software com al seu desenvolupament és la classe de risc (I, IIA, IIB o III) del software en funció de la seva usabilitat i grau d’error potencial que podem identificar en el funcionament, segons ha explicat Monferrer.
Segons s’estipula als nous reglaments, concretament la norma 11 de la Medical Device Regulation, d’entrada, el sofwtare com a MD està subjecte a la classe IIA, cosa que fa que hagi de passar per un procés de regulació de conformitat amb un organisme notificat . Els programaris que es classifiquen en IIB i III són aquells que tenen més risc d’impacte en la gestió del pacient. D’altra banda, els que es classifiquen a classe I (es donen en casos molt puntuals) tenen un control regulador molt lax i no han de passar per l’auditoria de l’organisme.
Tot i aquesta classificació, l’experta en regulació d’Asphalion ha indicat que s’han de tenir en compte moltes regles i subregles aplicables en funció de la tipologia i la finalitat de cada software i que s’ha de mirar cas per cas. Tot i això, ha afirmat que, en cas de dubte, sempre s’ha d’aplicar la regla i subregla més estricta que doni lloc a la classificació de risc més elevada.
Roadmap regulatori del software com a medical device, per fases
Tot software com a medical device té la potencialitat de resoldre una necessitat mèdica, però abans de convertir-se en un medical device ha de passar per un camí regulador complex i amb moltes fases intermèdies. En aquesta sessió, tots els assistents han pogut conèixer de primera mà el full de ruta que cal seguir per donar compliment a la regulació i el marc europeu que regeix aquestes tecnologies.
D’acord amb Dominique, el procés regulatori del software ve definit per 5 moments clau: disseny del programari, definició de la classe de risc, desenvolupament del producte (validació clínica), documentació tècnica i procediment d’avaluació de la conformitat.
Conceptualització i disseny del producte
Tal com ha explicat Monferrer, ja des de la configuració del producte, i a banda dels requisits de disseny (funcionalitat) i usabilitat (user-friendly), els fabricants de software han d’adoptar una mirada regulatòria per mostrar conformitat amb els requisits reguladors i normes tècniques (ISO).
Així mateix, és la fase en què es determina la classe de risc de la tecnologia, que, tal com s’ha esmentat anteriorment, ve marcada per les conseqüències d’un possible error del software en l’usuari. El producte també ha de complir amb especificacions quant a ciberseguretat i interoperabilitat, cosa que obliga els desenvolupadors a implementar plans de ciberseguretat.
A la sessió celebrada també s’ha posat la lupa sobre les principals diferències per discernir quan un programari es classifica com a medical device i, per tant, s’ha d’atenir a la Medical Device Regulation, i quan es tracta d’un producte de diagnòstic in vitro, governat per la In Vitro Diagnostic Regulation.
Tot i que tots dos tenen una finalitat mèdica i realitzen accions complexes de dades, el programari com a IVDR analitza una mostra biològica obtinguda del cos humà, incloses les donacions de sang i teixits, i la seva anàlisi ha de ser exviva.
Desenvolupament: verificació i validació clínica
A mig camí, els investigadors arriben al procés de desenvolupament del producte, on a banda de la verificació de la seva tecnologia, tot programari ha de passar per una avaluació clínica, per demostrar-ne la validesa amb dades clíniques, segons ha subratllat l’experta en medical devices d’Asphalion .
Documentació tècnica i comercialització
Un cop es duen a terme aquestes darreres fases del desenvolupament, té lloc la “compilació de documentació tècnica” (pack documental que descriu el producte: què és, ús previst, mecanisme d’acció o principi operatiu a través del qual s’assoleix la finalitat, procés de disseny i desenvolupament, verificació a tots els seus nivells i validació clínica final) per tal d’enviar-se a l’organisme notificat.
Un cop al mercat, el programari serà objecte de plans de monitorització i seguiment de postcomercialització per garantir la seva seguretat i eficiència. La regulació és un procés viu que obliga els i les investigadores a estar atents a tota la normativa vigent.
Tant si et trobes en fase de desenvolupament i avaluació clínica del teu programari com a medical device com si estàs a un pas de llançar-lo al mercat, necessites conèixer tota la regulació que envolta aquesta tecnologia. Aquesta sessió és una oportunitat per endinsar-se al complex camp regulatori. T’has perdut el B&L o vols tornar a veure la jornada? Et deixem la sessió gravada aquí.

